Home > Research > Research Areas > Miscellaneous
Highly interesting phenotypes unrelated to our core Research Areas are often identified among mutant mice outside of phenotypic screening and we pursue the mapping and identification of their causative mutations. In many instances the molecular analysis of such phenotypes, including coat color abnormalities, alopecia, and behavioral abnormalities, have resulted in significant biological insights. Several published examples are described below.
The seal phenotype, caused by a mutation of Col1a1, was an inducible seal-like gait resulting from grasping the loose skin at the back of the neck (“scruffing,” a routine method for immobilizing mice). The seal-like gait persisted for approximately eight days before returning to near normal. Spinal fracture, presumably resulting in hind limb paralysis, was found in mice with seal-like locomotion. In addition, homozygous seal mice had shortened limbs due to a reduction in the length of the long bones relative to that of wild type littermates.
Col1a1 encodes the type I collagen, α1 chain, two of which supercoil with one α2 chain to form a triple helix that is enzymatically cleaved to form mature type I collagen, the main structural component of mammalian bones. The seal mutation affected a critical residue of the intron 36 splice donor site, resulting in greatly reduced Col1a1 mRNA, and consequently reduced and aberrant collagen fibers in tibiae of seal homozygous mice. Unexpectedly, Col1a1seal mRNA from bone showed a normal pattern of splicing despite the presence of the donor splice site mutation. Further investigation suggested that a putative intronic splicing enhancer in intron 25 may function redundantly with the intron 36 splice donor site.
The helical domain of the α1 chain of type I collagen is encoded by 43 out of 51 total exons; these 43 exons code for the repeating amino acid sequence Gly-X-Y. The presence of a glycine every third residue is critical for the formation of the mature type I collagen triple helix; thus frameshifts or premature termination caused by aberrant splicing can hinder proper type I collagen synthesis. Our findings revealed a previously unknown mechanism for splicing rescue, which may represent an adaptation that evolved to enforce proper splicing of collagen mRNAs.
The seal phenotype and causative Col1a1 mutation are reported in (Tabeta et al. Sci.Rep. 7, 11717).
Study of the Gk5 mutation toku revealed a skin-specific regulatory mechanism for cholesterol biosynthesis dependent on glycerol kinase 5 (GK5) and the sterol regulatory element-binding proteins (SREBPs). The toku phenotype was characterized by delayed hair growth and progressive hair loss due to defective hair follicle morphogenesis and maintenance, impaired progression through the hair cycle, and degeneration of the hair follicle. Excessive accumulation of lipids (cholesterol, triglycerides, and ceramides) occurred in the skin of toku homozygous mice. These defects were attributed to a null mutation of GK5; mice with normal expression levels of a kinase-inactive GK5 also showed similar defects. The hair loss of toku homozygous mice was rescued by topical treatment with simvastatin, an inhibitor of HMG-CoA reductase, which catalyzes the rate-limiting step of cholesterol biosynthesis. GK5 expression was found to be restricted to the sebaceous glands of the skin. These findings suggested that GK5 phosphorylates substrate(s) to regulate cholesterol biosynthesis in the skin.
Further investigation showed that GK5 binds to and inhibits SREBPs, key transcription factors that promote cholesterol biosynthesis and homeostasis by stimulating the transcription of cholesterol synthesis enzymes. Thus, in Gk5toku/toku mice, transcriptionally active SREBPs accumulated in the skin, but not in the liver; they were localized to the nucleus and led to elevated lipid synthesis and subsequent hair growth defects. The relevant substrate(s) phosphorylated by GK5 to regulate cholesterol biosynthesis in the skin remain unknown, but neither SREBPs nor glycerol are believed to be critical substrates. Further work is necessary to elucidate the mechanistic link between GK5 and SREBPs, which may entail crosstalk between the glycerol metabolism pathway and the cholesterol biosynthesis pathway.
The toku phenotype and causative Gk5 mutation are reported in (Zhang et al. Proc.Natl.Acad.Sci.U.S.A. 114, E5197-E5206).
A hair loss phenotype called mask (Figure 1) led to the discovery of a fundamental regulatory mechanism for intestinal iron absorption. Because no regulated mechanism exists for the excretion of excess iron from the body, body iron levels are controlled through regulated absorption of iron in the small intestine (Figure 2A). The peptide hormone hepcidin, encoded by Hamp, is a major systemic iron regulator secreted by the liver to limit intestinal iron absorption. Thus, Hamp expression is induced by replete body iron stores and inhibited by anemia and hypoxia. Positive regulation of Hamp expression involves hemochromatosis protein (HFE), transferrin receptor 2 (TFR2), hemojuvelin (HJV), and the transcription factor SMAD4 (Figure 2B). Study of the mask phenotype revealed that TMPRSS6 is an essential component of a pathway that detects iron deficiency and blocks Hamp transcription, permitting enhanced dietary iron absorption (Figure 2B).
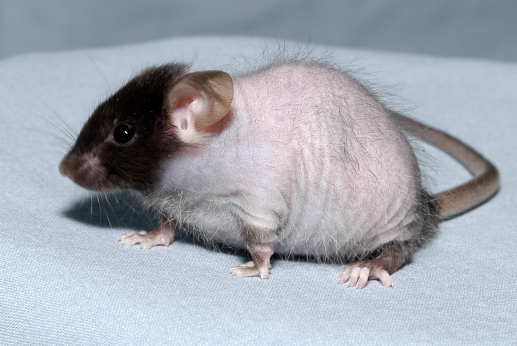

The recessive mask mutant phenotype emerged as a visible variant among G3 mice. Mask mice initially grew a normal, full coat of hair by postnatal day 10, but then gradually lost hair from the trunk beginning at P13 until they were nude by P28; however, facial hair was preserved (Figure 1). Mask mutant mice showed evidence of iron deficiency including microcytic anemia, low plasma iron levels, and depleted tissue iron stores, but no occult blood loss. Intestinal iron absorption by mask mice measured after two weeks on an iron-deficient diet was approximately half that measured in wild type mice. Moreover, iron-deprived mask homozygotes expressed approximately 8-fold higher levels of hepcidin-encoding mRNA compared to iron-deprived control mice, consistent with insensitivity to low iron stores and consequent failure to suppress Hamp expression. The mask phenotype was attributed to a mutation in Tmprss6, encoding a transmembrane serine protease of unknown function, also called matriptase-2. TMPRSS6 overexpression inhibited Hamp expression induced by activating stimuli such as interleukin-1α (IL-1α), IL-6, bone morphogenetic protein 2 (BMP2), or BMP4, or by expression of hemojuvelin or SMAD1. Although the protease activity of TMPRSS6 is necessary for Hamp suppression through the BMP/SMAD pathway, the mechanism is not yet fully understood. Mutations in TMPRSS6 in humans cause iron-refractory iron deficiency anemia (IRIDA) (Finberg et al. Nat.Genet. 40, 569-571).
The mask phenotype and causative Tmprss6 mutation are reported in (Du et al. Science. 320, 1088-1092).
The aoba mutation in the type IV collagen α4 chain encoded by Col4a4 was found to be responsible for spontaneous death of homozygous mice around six months of age due to end-stage renal disease (ESRD) and kidney failure. By two months of age, aoba mutant mice had proteinuria, hematuria, and leukocytes in the urine; at an advanced stage of illness, they had high levels of blood urea nitrogen (BUN) and creatinine, and small, pale kidneys with focal glomerusclerosis. The type IV collagen α3, α4, and α5 chains form a heterotrimer; subsequent dimerization via the NC1 domain, and tetermerization via the 7S domain result in the formation of a type IV collagen network that is critical to the function of the kidney glomerular basement membrane in filtration of plasma. Since the α3, α4, and α5 chains form a trimeric molecule, a mutation in any of these chains usually results in complete absence of the α3 α4 α5 collagen IV network. As in mice, mutations of COL4A3, COL4A4, or COL4A5 in humans have been shown to cause Alport syndrome and/or autosomal dominant benign familial hematuria, inherited kidney diseases involving the glomerular basement membrane.
The aoba phenotype and causative Col4a4 mutation are reported in (Arnold et al. Genetics. 187, 633-641). This is one of the first reports of the use of whole genome sequencing together with the quick mapping technique bulk segregation analysis to identify a causative mutation.
Publications
- Tabeta, K., Du, X., Arimatsu, K., Yokoji, M., Takahashi, N., Amizuka, N., Hasegawa, T., Crozat, K., Maekawa, T., Miyauchi, S., Matsuda, Y., Ida, T., Kaku, M., Hoebe, K., Ohno, K., Yoshie, H., Yamazaki, K., Moresco, E. M. Y., and Beutler, B. (2017) An ENU-Induced Splice Site Mutation of Mouse Col1a1 Causing Recessive Osteogenesis Imperfecta and Revealing a Novel Splicing Rescue. Sci Rep. 7, 11717-017-10343-9.
- Zhang, D., Tomisato, W., Su, L., Sun, L., Choi, J. H., Zhang, Z., Wang, K. W., Zhan, X., Choi, M., Li, X., Tang, M., Castro-Perez, J. M., Hildebrand, S., Murray, A. R., Moresco, E. M. Y., and Beutler, B. (2017) Skin-Specific Regulation of SREBP Processing and Lipid Biosynthesis by Glycerol Kinase 5. Proc Natl Acad Sci U S A. 114, E5197-E5206.
- Rutschmann, S., Crozat, K., Li, X., Du, X., Hanselman, J. C., Shigeoka, A. A., Brandl, K., Popkin, D. L., McKay, D. B., Xia, Y., Moresco, E. M., and Beutler, B. (2012) Hypopigmentation and Maternal-Zygotic Embryonic Lethality Caused by a Hypomorphic mbtps1 Mutation in Mice. G3 (Bethesda). 2, 499-504.
- Siggs, O. M., Cruite, J. T., Du, X., Rutschmann, S., Masliah, E., Beutler, B., and Oldstone, M. B. (2012) Disruption of Copper Homeostasis due to a Mutation of Atp7a Delays the Onset of Prion Disease. Proc Natl Acad Sci U S A.
- Arnold, C. N., Xia, Y., Lin, P., Ross, C., Schwander, M., Smart, N. G., Muller, U., and Beutler, B. (2011) Rapid Identification of a Disease Allele in Mouse through Whole Genome Sequencing and Bulk Segregation Analysis. Genetics. 187, 633-641.
- Krebs, P., Fan, W., Chen, Y. H., Tobita, K., Downes, M. R., Wood, M. R., Sun, L., Li, X., Xia, Y., Ding, N., Spaeth, J. M., Moresco, E. M., Boyer, T. G., Lo, C. W., Yen, J., Evans, R. M., and Beutler, B. (2011) Lethal Mitochondrial Cardiomyopathy in a Hypomorphic Med30 Mouse Mutant is Ameliorated by Ketogenic Diet. Proc Natl Acad Sci U S A. 108, 19678-19682.
- Krieg, L., Milstein, O., Krebs, P., Xia, Y., Beutler, B., and Du, X. (2011) Mutation of the Gastric Hydrogen-Potassium ATPase Alpha Subunit Causes Iron-Deficiency Anemia in Mice. Blood. 118, 6418-6425.
- Siggs, O. M., Schnabl, B., Webb, B., and Beutler, B. (2011) X-Linked Cholestasis in Mouse due to Mutations of the P4-ATPase ATP11C. Proc Natl Acad Sci U S A. 108, 7890-7895.
- Blasius, A. L., Brandl, K., Crozat, K., Xia, Y., Khovananth, K., Krebs, P., Smart, N. G., Zampolli, A., Ruggeri, Z. M., and Beutler, B. A. (2009) Mice with Mutations of Dock7 have Generalized Hypopigmentation and White-Spotting but show Normal Neurological Function. Proc Natl Acad Sci U S A. 106, 2706-2711.
- Du, X., She, E., Gelbart, T., Truksa, J., Lee, P., Xia, Y., Khovananth, K., Mudd, S., Mann, N., Moresco, E. M., Beutler, E., and Beutler, B. (2008) The Serine Protease TMPRSS6 is Required to Sense Iron Deficiency. Science. 320, 1088-1092.